Le colobome représente une malformation congénitale rare de l’œil qui affecte environ 1 naissance sur 10 000. Cette anomalie résulte d’un défaut de fermeture de la fissure embryonnaire au cours du développement fœtal, créant une absence de tissu dans une ou plusieurs structures oculaires. Bien que souvent méconnue du grand public, cette pathologie peut avoir des implications visuelles significatives selon les structures affectées. La compréhension de cette malformation s’avère essentielle pour les professionnels de santé comme pour les familles concernées, car elle nécessite une prise en charge précoce et adaptée. Le pronostic visuel dépend largement de la localisation et de l’étendue de la malformation, pouvant varier d’un simple défaut esthétique à une déficience visuelle majeure.
Définition anatomique et classification embryologique du colobome oculaire
Le terme « colobome » dérive du grec ancien « koloboma » signifiant « mutiler » ou « amputer », une étymologie qui illustre parfaitement la nature de cette malformation caractérisée par l’absence de tissu oculaire. Cette anomalie congénitale résulte d’un processus embryologique complexe impliquant la fermeture imparfaite de la fissure choroïdienne pendant le développement précoce de l’œil. La formation normale de l’œil nécessite une orchestration précise de mécanismes moléculaires et cellulaires qui, lorsqu’ils sont perturbés, peuvent conduire à diverses formes de colobomes.
Anomalies de fermeture de la fissure choroïdienne embryonnaire
La fissure choroïdienne embryonnaire se forme vers la cinquième semaine de gestation et doit normalement se refermer complètement entre la sixième et la septième semaine de développement fœtal. Cette fissure permet initialement le passage de l’artère hyaloïde et la formation du pédicule optique. L’échec de cette fermeture, même partiel, entraîne la persistance d’une ouverture qui se manifeste sous forme de colobome. Ce processus implique des cascades de signalisation complexes régulées par des facteurs de transcription spécifiques, notamment les gènes de la famille PAX.
Les mécanismes moléculaires sous-jacents font intervenir la migration cellulaire, la différenciation tissulaire et l’apoptose programmée. Quand ces processus sont altérés, que ce soit par des mutations génétiques ou des facteurs environnementaux, la fermeture normale de la fissure ne peut s’effectuer correctement. Cette anomalie peut affecter différentes structures selon le moment précis où survient la perturbation développementale.
Classification topographique : colobomes iris, rétine, choroïde et nerf optique
La classification anatomique des colobomes repose sur la localisation des structures affectées. Le colobome irien représente la forme la plus visible, créant une déformation caractéristique en « trou de serrure » de la pupille. Cette anomalie peut être complète, affectant toute l’épaisseur de l’iris, ou incomplète, ne touchant qu’une partie de cette structure. L’impact fonctionnel reste généralement limité, bien qu’une photophobie puisse se manifester en cas de colobome étendu.
Les colobomes choriorétiniens présentent des implications visuelles bien plus sérieuses. Ces malformations affectent les couches profondes de l’œil responsables de la perception visuelle et peuvent entraîner des déficits visuels permanents. Le colobome du nerf optique, souvent associé aux formes
du segment postérieur, peut conduire à une baisse d’acuité visuelle sévère et à des anomalies du champ visuel. Enfin, certains colobomes intéressent le cristallin (colobome cristallinien) ou les corps ciliaires, souvent en regard d’un colobome irien, et s’intègrent dans un spectre plus large de malformations oculaires congénitales. Dans la pratique, un même patient peut donc présenter plusieurs types de colobomes associés, ce qui explique la grande variabilité du retentissement fonctionnel d’un colobome oculaire.
Différenciation entre colobomes typiques et atypiques selon leur localisation
Sur le plan anatomique, on distingue les colobomes typiques et les colobomes atypiques. Les colobomes typiques sont situés dans le quadrant inférieur (souvent inférieur et légèrement nasal), en continuité avec la zone de la fissure choroïdienne embryonnaire. Ils concernent classiquement l’iris, la rétine, la choroïde ou la papille optique, avec un aspect en entaille ou en excavation blanche bien limitée. Les colobomes atypiques, eux, se développent en dehors de cette zone embryonnaire, par exemple au pôle maculaire, et répondent à d’autres mécanismes de dysgénésie rétinienne.
Cette distinction n’est pas qu’académique : elle permet d’orienter le diagnostic étiologique et d’anticiper certaines complications. Un colobome typique chorio-rétinien inférieur sera, par exemple, plus volontiers associé à un risque de décollement de rétine sur ses bords amincis, tandis qu’un colobome maculaire isolé entraîne surtout une baisse centrale d’acuité visuelle sans forcément fragiliser la périphérie rétinienne. Pour vous, clinicien ou parent, comprendre si le colobome est typique ou atypique aide à mieux interpréter les examens d’imagerie et à planifier la surveillance.
Les colobomes atypiques de la région maculaire peuvent mimer d’autres pathologies telles que la maladie de Leber, une cicatrice de toxoplasmose congénitale ou certaines dystrophies rétiniennes. L’analyse détaillée en tomographie par cohérence optique (OCT) et angiographie fluorescéinique est alors essentielle pour visualiser l’absence de couches rétiniennes normales au sein de l’excavation colobomateuse. En pratique, le colobome oculaire se situe aujourd’hui sur un continuum de malformations rétiniennes, allant de simples irrégularités structurelles à des déficits profonds de tissus neuro-sensoriels.
Spectre des malformations associées au syndrome CHARGE et PAX6
Le colobome s’intègre fréquemment dans un cadre syndromique plus large, dont le plus connu est le syndrome CHARGE. Cet acronyme anglais résume les principales anomalies : Coloboma oculaire, Heart defects (malformations cardiaques), Atresia choanae, Retardation of growth and development, anomalies Genitourinary et Ear abnormalities. Dans ce contexte, le colobome est souvent bilatéral, intéressant l’iris et la rétine, et s’associe à une microphtalmie. Pour les familles, il est crucial de savoir qu’un colobome n’est pas seulement un problème oculaire : il peut être la porte d’entrée vers le diagnostic d’un syndrome multisystémique.
Le gène CHD7 est le principal gène impliqué dans le syndrome CHARGE, mais d’autres gènes de développement oculaire, comme PAX6, interviennent également dans le spectre des malformations colobomateuses. PAX6 est considéré comme un gène maître de l’œil : des mutations peuvent provoquer aniridie, colobome, microphtalmie ou autres dysgénésies du segment antérieur. On peut comparer son rôle à celui d’un chef d’orchestre : lorsqu’il est défaillant, l’ensemble de la « symphonie » du développement oculaire est désorganisé.
De façon plus large, le spectre PAX6 associe anomalies de la cornée, de l’iris, du cristallin et de la rétine, avec des profils visuels très variables. Certains enfants porteront un colobome irien relativement discret, principalement gênant par la photophobie, tandis que d’autres présenteront une lésion maculaire colobomateuse responsable d’une malvoyance sévère. Dans ces situations, une évaluation génétique spécialisée est recommandée afin d’identifier précisément le gène en cause et de proposer un conseil génétique adapté à la famille.
Étiologie génétique et facteurs de risque embryonnaires
La genèse d’un colobome oculaire résulte en grande partie de facteurs génétiques, parfois associés à des influences environnementales au cours de la grossesse. Les progrès de la génétique moléculaire ont permis d’identifier plusieurs gènes clés impliqués dans la fermeture de la fissure embryonnaire, mais aussi dans la différenciation des différentes structures de l’œil. Comprendre ces mécanismes permet non seulement de mieux expliquer la survenue du colobome aux familles, mais aussi d’évaluer le risque de récurrence lors d’une nouvelle grossesse.
Mutations des gènes PAX2, PAX6 et CHD7 dans la genèse colobomateuse
Les gènes PAX2 et PAX6 appartiennent à la famille des facteurs de transcription PAX, essentiels dans le développement des yeux, des reins et du système nerveux central. Des mutations de PAX2 sont retrouvées notamment dans le syndrome rein-colobome, associant anomalies rénales congénitales et colobome chorio-rétinien ou papillaire. Dans ces cas, l’examen ophtalmologique joue parfois un rôle clé de dépistage d’une pathologie rénale sous-jacente. Le gène PAX6, quant à lui, est impliqué dans une large gamme de malformations oculaires, de l’aniridie aux colobomes complexes.
Le gène CHD7, déjà mentionné dans le syndrome CHARGE, code pour une protéine intervenant dans la régulation de la chromatine et l’expression de nombreux autres gènes du développement. Des variants pathogènes de CHD7 perturbent des cascades entières de différenciation cellulaire, ce qui explique le caractère polysystémique (œil, oreille, cœur, système nerveux) des atteintes. On peut comparer cette régulation à un système de dominos : lorsque l’un des éléments tombe (mutation), l’ensemble de la chaîne de développement embryonnaire est impactée.
Dans la pratique clinique, l’identification d’une mutation de PAX2, PAX6 ou CHD7 se fait par séquençage ciblé ou par panels de gènes dédiés aux malformations oculaires congénitales. Pour les parents, cette démarche permet d’obtenir une explication étiologique, de préciser le mode de transmission et de discuter le risque pour une future grossesse. Même si cette information ne modifie pas le colobome déjà constitué, elle influence directement la stratégie de suivi familial et la prévention.
Syndrome oculo-auriculo-vertébral de goldenhar et colobomes associés
Le syndrome de Goldenhar, ou syndrome oculo-auriculo-vertébral, est une autre entité dans laquelle des colobomes peuvent être observés. Il s’agit d’un trouble du développement des arcs branchiaux, se traduisant par des anomalies faciales asymétriques, des malformations auriculaires (microtie, oreilles malformées) et des défauts vertébraux. Sur le plan oculaire, on retrouve parfois des dermoïdes limbiques, une microphtalmie et des colobomes palpébraux ou iriens. Dans ce contexte, le colobome n’est qu’une composante d’un tableau cranio-facial plus global.
Le colobome de la paupière, typique de ce spectre, correspond à une encoche congénitale du bord palpébral, le plus souvent au niveau de la paupière supérieure ou inférieure, exposant partiellement la cornée. Ce défaut peut sembler mineur mais entraîne un risque de kératite d’exposition et de séquelles cornéennes définitives si une prise en charge précoce n’est pas mise en place. Chez le nourrisson, vous comprendrez qu’une œil mal protégé par la paupière est plus vulnérable aux agressions mécaniques et à la sécheresse.
Le mécanisme précis de ce syndrome reste multifactoriel, associant probablement des facteurs génétiques encore imparfaitement identifiés et des influences environnementales. Pour le clinicien, l’identification d’un colobome palpébral ou irien dans un contexte de dysmorphie faciale doit faire évoquer un syndrome de Goldenhar et conduire à un bilan complet (imagerie vertébrale, examen ORL, évaluation audiologique). Une prise en charge multidisciplinaire précoce limite alors le risque de complications visuelles et fonctionnelles à long terme.
Facteurs tératogènes maternels : alcool, isotrétinoïne et infections TORCH
Au-delà des gènes, plusieurs facteurs environnementaux pendant la grossesse augmentent le risque de malformations oculaires, y compris de colobomes. L’alcool, responsable du syndrome d’alcoolisation fœtale, est bien documenté comme tératogène majeur, avec possibilité de dysmorphie faciale, retard de croissance et anomalies oculaires. L’isotrétinoïne, dérivé de la vitamine A utilisé dans le traitement de l’acné sévère, est également un tératogène puissant, formellement contre-indiqué pendant la grossesse en raison de son association avec des malformations cranio-faciales et oculaires.
Les infections maternelles regroupées sous l’acronyme TORCH (toxoplasmose, rubéole, cytomégalovirus, herpès et autres agents) peuvent aussi perturber le développement oculaire in utero. Elles sont plus classiquement associées à des choriorétinites cicatricielles, mais des défauts de différenciation rétinienne pouvant mimer ou coexister avec des colobomes ont été décrits. Comme pour d’autres anomalies congénitales, la période critique se situe au premier trimestre, lorsque les structures oculaires se forment de manière très rapide.
La prévention de ces facteurs de risque est un axe majeur de santé publique. Informer les femmes en âge de procréer de la nécessité d’éviter l’alcool et certains médicaments tératogènes, assurer une couverture vaccinale antirubéole correcte et proposer un suivi obstétrical adapté sont des mesures concrètes que l’on peut mettre en œuvre. Même si tous les colobomes ne sont pas évitables, réduire l’exposition aux principaux tératogènes contribue à diminuer la fréquence des malformations oculaires graves.
Hérédité autosomique dominante dans le syndrome colobome-microphtalmie
Certains tableaux cliniques associent colobome et microphtalmie dans un cadre familial, avec un mode de transmission souvent autosomique dominant. Cela signifie qu’une seule copie altérée du gène concerné suffit à transmettre la maladie à la descendance, avec un risque de 50 % pour chaque grossesse. Toutefois, l’expressivité est variable : au sein d’une même famille, certains membres présenteront un colobome sévère bilatéral, d’autres seulement une microphtalmie discrète ou un colobome irien isolé.
Des gènes comme SOX2, OTX2 ou VSX2 peuvent être impliqués dans ces syndromes colobome-microphtalmie, mais le paysage génétique reste en cours d’exploration. Dans ce contexte, le recours à la génétique moléculaire est particulièrement utile pour confirmer le mécanisme autosomique dominant et offrir un conseil génétique fiable. Vous vous interrogez sur le risque pour un futur enfant lorsque l’un des parents est porteur d’un colobome ? Un avis en consultation de génétique clinique est alors fortement recommandé.
Pour les familles concernées, la connaissance du mode de transmission permet également d’anticiper un dépistage néonatal ciblé chez les nouveau-nés à risque. Un examen ophtalmologique précoce, dès les premières semaines de vie, permet en effet de détecter rapidement un colobome et de mettre en place la rééducation visuelle et la correction optique nécessaires pendant la période de plasticité cérébrale maximale.
Manifestations cliniques et diagnostic ophtalmologique différentiel
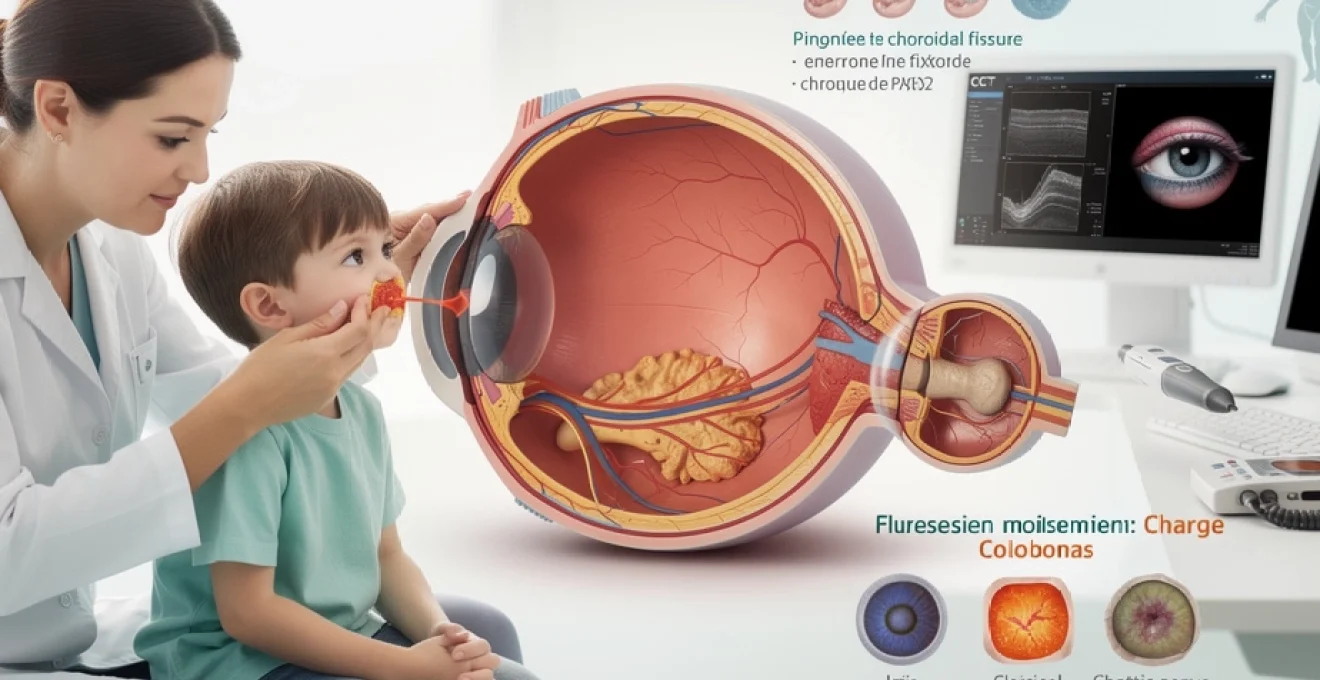
Les manifestations cliniques du colobome oculaire sont extrêmement variées, allant d’une simple anomalie esthétique de la pupille à une atteinte visuelle profonde avec malvoyance. L’intensité des symptômes dépend de la localisation, de l’étendue de la malformation et des structures associées touchées (rétine, nerf optique, cristallin). Le diagnostic est avant tout clinique, mais il est largement complété aujourd’hui par des techniques d’imagerie moderne qui permettent de préciser la topographie et la profondeur des lésions.
Techniques d’imagerie : OCT, échographie oculaire et angiographie fluorescéinique
La tomographie par cohérence optique (OCT) est devenue un examen de référence dans l’exploration des colobomes rétiniens et maculaires. Elle fournit des coupes en haute résolution des différentes couches de la rétine, mettant en évidence l’absence ou la désorganisation des tissus au sein de la zone colobomateuse. L’OCT permet également de détecter d’éventuelles complications, comme l’apparition de néovaisseaux choroïdiens sur les bords du colobome, avant même que les symptômes n’apparaissent. Pour le patient, l’examen est rapide, indolore et non invasif.
L’échographie oculaire en mode B est particulièrement utile lorsque les milieux sont opaques (cataracte dense, hémorragie vitréenne) ou en cas de microphtalmie importante. Elle permet de visualiser l’architecture globale du globe oculaire, d’identifier un colobome profond du pôle postérieur et de rechercher un décollement de rétine associé. L’analogie avec un sonar est souvent utilisée : l’échographie envoie des ondes qui se réfléchissent sur les structures internes, restituant une image de leur relief.
L’angiographie fluorescéinique et, plus récemment, l’angiographie au vert d’indocyanine ou par OCT-angiographie, apportent des informations sur la vascularisation rétinienne et choroïdienne autour du colobome. Elles sont particulièrement indiquées en cas de baisse visuelle récente, pour rechercher une complication néovasculaire traitable par injections intravitréennes. Ces examens complètent l’ophtalmoscopie indirecte et constituent un outil précieux pour le suivi à long terme, notamment chez l’adulte jeune avec colobome chorio-rétinien.
Évaluation de l’acuité visuelle et déficits campométriques spécifiques
L’évaluation fonctionnelle repose d’abord sur la mesure de l’acuité visuelle, adaptée à l’âge du patient. Chez le nourrisson, on utilise des méthodes comportementales ou des tests de regard préférentiel ; chez l’enfant plus grand et l’adulte, on a recours aux échelles classiques (Monoyer, LogMAR, etc.). L’acuité visuelle peut être étonnamment préservée dans les colobomes iriens isolés, alors qu’elle est souvent très réduite dans les colobomes maculaires ou du nerf optique. Tout l’enjeu est d’identifier ce potentiel visuel pour mettre en place une correction optique et une rééducation adaptées.
Les déficits campométriques (anomalies du champ visuel) sont caractéristiques des colobomes du pôle postérieur. Un colobome infero-rétinien créera généralement un scotome supérieur, parfois étendu, pouvant gêner la lecture ou les déplacements. Les examens de périmétrie statique ou cinétique (type Goldmann) permettent de cartographier ces pertes de sensibilité. Imaginez le champ visuel comme une carte géographique : le colobome y dessine une « zone blanche » permanente, que le cerveau pourra parfois partiellement compenser par des stratégies d’exploration visuelle.
Il est important d’expliquer au patient que ces déficits de champ visuel sont structurels et ne peuvent pas être « récupérés » par un traitement médical ou chirurgical. En revanche, un accompagnement en basse vision, avec des conseils de réadaptation et parfois des aides optiques spécifiques, permet souvent d’améliorer la qualité de vie au quotidien. Une évaluation orthoptique régulière aide également à dépister et traiter une éventuelle amblyopie chez l’enfant.
Diagnostic différentiel avec décollement rétinien et staphylome postérieur
Sur le plan ophtalmoscopique, certains colobomes chorio-rétiniens peuvent prêter à confusion avec d’autres affections du fond d’œil. Le décollement de rétine congénital ou tractionnel présente parfois des zones d’atrophie rétinienne et de soulèvement qui évoquent un colobome. L’angiographie et surtout l’OCT permettent en général de trancher, en montrant d’une part la présence de liquide sous-rétinien dans le décollement, d’autre part l’absence de tissu normal dans le colobome. Le bon diagnostic conditionne la prise en charge, le décollement pouvant bénéficier d’un traitement chirurgical spécifique.
Le staphylome postérieur, qui correspond à une ectasie sclérale localisée, notamment dans les myopies fortes, peut lui aussi simuler une excavation colobomateuse au pôle postérieur. Toutefois, la topographie et les contours de la lésion diffèrent : le staphylome a des bords plus progressifs et une extension souvent plus diffuse. L’imagerie multimodale (OCT, échographie, parfois IRM orbitaire) aide à distinguer ces entités. Pour le praticien, garder en tête ce diagnostic différentiel est essentiel afin de ne pas attribuer à tort une lésion acquise à une malformation congénitale.
D’autres affections, telles que les cicatrices de toxoplasmose congénitale, certaines dystrophies rétiniennes ou les séquelles d’inflammations intraoculaires, peuvent également mimer un colobome atypique. Un interrogatoire précis (antécédents maternels, contexte périnatal) et une évaluation systémique orientent alors vers la bonne étiologie. Dans le doute, l’avis d’un spécialiste en rétine médicale ou en maladies rares ophtalmiques est préférable.
Dépistage néonatal systématique par ophtalmoscopie indirecte
Le colobome oculaire, notamment lorsqu’il est bilatéral et associé à une microphtalmie, peut être repéré très tôt, parfois dès la maternité. Un dépistage néonatal systématique par ophtalmoscopie indirecte n’est pas encore généralisé dans tous les pays, mais un examen attentif du reflet pupillaire et de la morphologie de l’iris fait partie de l’examen clinique de base du nouveau-né. En cas d’anomalie évidente (pupille en trou de serrure, asymétrie des globes, reflet rouge anormal), un avis ophtalmologique spécialisé rapide est indispensable.
Chez les enfants appartenant à des familles à risque (antécédent de colobome, syndrome génétique connu), un examen ophtalmologique complet est recommandé dans les premières semaines de vie. L’ophtalmoscopie indirecte, parfois sous sédation légère ou anesthésie générale selon l’âge et la coopération de l’enfant, permet alors de cartographier précisément l’étendue des lésions. Plus le diagnostic est précoce, plus la prise en charge de l’amblyopie, la correction optique et la mise en place d’aides à la basse vision pourront être efficaces.
Vous vous demandez si un colobome peut passer inaperçu à la naissance ? C’est possible pour de petites formes iriens ou des colobomes rétiniens périphériques, asymptomatiques au début. D’où l’importance de contrôles ophtalmologiques systématiques chez l’enfant, notamment en cas de strabisme, de retard d’acquisition visuelle ou de plaintes de photophobie. Une détection précoce reste le meilleur levier pour optimiser le pronostic fonctionnel.
Complications oculaires et systémiques associées
Le colobome n’est pas une anomalie figée sans conséquence : au fil du temps, il peut s’accompagner de complications oculaires parfois sévères. Sur le plan rétinien, le risque majeur est le décollement de rétine, favorisé par la fragilité des bords colobomateux et d’éventuelles tractions vitréennes. Des néovaisseaux choroïdiens peuvent aussi se développer à la périphérie du colobome, entraînant des hémorragies et une baisse visuelle brutale. Ces complications nécessitent un suivi régulier et une prise en charge rapide (laser, injections intravitréennes, chirurgie).
Au niveau de l’iris, un colobome large induit souvent une photophobie importante, gênante au quotidien, et peut être à l’origine d’un astigmatisme significatif. Le cristallin, quant à lui, est exposé à un risque accru de cataracte, notamment dans le cadre de syndromes comme l’aniridie ou le syndrome CHARGE. Des ectopies cristalliniennes (déplacement du cristallin) peuvent également être observées, compliquant la correction optique. Le colobome du nerf optique est, de son côté, associé à des déficits visuels irréversibles par atteinte directe des fibres optiques.
Les complications ne se limitent pas à l’œil. Dans les formes syndromiques, en particulier le syndrome CHARGE, les répercussions systémiques sont multiples : malformations cardiaques, troubles auditifs, difficultés respiratoires, anomalies uro-génitales. Ces co-morbidités influencent directement la qualité de vie et le développement global de l’enfant. C’est pourquoi la découverte d’un colobome doit toujours faire rechercher d’autres anomalies organiques par un bilan pluridisciplinaire (cardiologie, ORL, néphrologie, génétique).
Sur le plan fonctionnel, les enfants atteints de colobome sont plus exposés à l’amblyopie (œil paresseux), au strabisme et parfois au nystagmus (mouvements oculaires involontaires). Ces troubles, s’ils ne sont pas pris en charge tôt, peuvent aggraver la malvoyance liée à la malformation anatomique initiale. L’impact psychologique et social ne doit pas être sous-estimé, surtout chez les adolescents pour qui l’aspect esthétique de l’œil peut être source de complexe. Un accompagnement psychologique et un soutien scolaire adapté font partie intégrante de la prise en charge globale.
Approches thérapeutiques chirurgicales et correction optique
Il n’existe pas à proprement parler de « chirurgie du colobome » capable de recréer le tissu oculaire manquant. En revanche, plusieurs approches thérapeutiques visent à corriger les conséquences fonctionnelles et esthétiques de la malformation. Le traitement est toujours personnalisé, en fonction du type de colobome, de l’âge du patient et de la présence de complications associées. L’objectif est double : protéger les structures oculaires encore fonctionnelles et optimiser le potentiel visuel restant.
Dans les colobomes iriens, la correction optique joue un rôle central. Une paire de lunettes correctement adaptée permet de corriger l’astigmatisme induit et d’éviter l’amblyopie réfractive chez l’enfant. Des filtres solaires ou des verres fortement teintés sont souvent nécessaires pour limiter la photophobie. Chez certains patients, des lentilles de contact prothétiques, reproduisant artificiellement un iris complet, offrent à la fois un confort visuel et une amélioration esthétique appréciable. Vous seriez surpris de la qualité cosmétique actuelle de ces dispositifs, quasi indiscernables d’un iris normal.
Sur le plan chirurgical, des techniques de plastie irienne permettent parfois de réduire l’ouverture colobomateuse, en rapprochant les bords de l’iris ou en implantant un diaphragme artificiel. Ces interventions sont délicates et réservées à des cas sélectionnés, chez l’adulte ou l’adolescent, lorsque la gêne fonctionnelle ou esthétique est majeure. Pour les colobomes associés à une cataracte significative, une chirurgie du cristallin avec implantation d’un implant intraoculaire est envisageable, mais elle nécessite une planification soigneuse compte tenu de l’anatomie particulière de l’œil colobomateux.
En cas de décollement de rétine sur colobome chorio-rétinien, une chirurgie vitréo-rétinienne spécialisée est indiquée, combinant parfois indentation sclérale, vitrectomie et tamponnement interne. Le pronostic anatomique est souvent bon si la prise en charge est précoce, mais le pronostic fonctionnel dépend étroitement de l’intégrité des zones rétiniennes non colobomateuses. Des traitements par laser ou par injections intravitréennes (anti-VEGF) peuvent également être proposés en présence de néovaisseaux choroïdiens périphériques du colobome.
Enfin, la rééducation orthoptique et les solutions de basse vision complètent l’arsenal thérapeutique. L’orthoptie vise à traiter l’amblyopie, le strabisme et à améliorer la coordination oculo-manuelle, en particulier chez l’enfant. Les aides de basse vision (loupes électroniques, systèmes télescopiques, filtres contrastés) sont précieuses pour les patients dont l’acuité visuelle reste limitée malgré toutes les corrections. Elles permettent de maintenir l’autonomie dans les activités quotidiennes, la lecture et la scolarité ou la vie professionnelle.
Pronostic visuel et prise en charge multidisciplinaire spécialisée
Le pronostic visuel du colobome est très variable et dépend principalement de la localisation et de l’étendue de la malformation. Un colobome irien isolé, sans atteinte rétinienne ni nerveuse, est souvent compatible avec une vision quasi normale, sous réserve d’une correction optique adaptée et d’un suivi régulier. À l’inverse, un colobome maculaire bilatéral ou un colobome étendu du nerf optique entraînent le plus souvent une malvoyance sévère, parfois dès la petite enfance. Pour les familles, il est important de comprendre que l’on ne peut pas « réparer » le tissu manquant, mais que l’on peut optimiser au maximum les capacités restantes.
La prise en charge idéale est multidisciplinaire, associant ophtalmologistes, orthoptistes, opticiens spécialisés en basse vision, mais aussi pédiatres, ORL, cardiologues et généticiens dans les formes syndromiques. Ce travail en réseau permet de coordonner les interventions, de planifier les contrôles et d’ajuster le projet de soins au développement de l’enfant. Par exemple, un enfant atteint de syndrome CHARGE bénéficiera à la fois d’un suivi ophtalmologique rapproché, d’un appareillage auditif précoce et d’un accompagnement psychomoteur pour favoriser son autonomie.
Sur le plan éducatif et social, l’orientation vers des services d’accompagnement pour enfants ou adultes malvoyants peut être proposée. Ceux-ci offrent un soutien pour l’adaptation scolaire, la formation professionnelle, l’accès aux aides techniques et la reconnaissance du handicap visuel le cas échéant. Vous vous demandez si un enfant colobomateux pourra suivre une scolarité ordinaire ? Dans de nombreux cas, la réponse est oui, à condition de mettre en place les aménagements nécessaires (place en classe, supports agrandis, éclairage adapté).
À plus long terme, les avancées en thérapie génique, en médecine régénérative et en implants rétiniens pourraient ouvrir de nouvelles perspectives pour certaines formes de colobomes, même si nous n’en sommes encore qu’aux premiers stades de recherche. En attendant, le meilleur atout reste un diagnostic précoce, un suivi spécialisé régulier et une information claire des familles sur les possibilités et les limites des traitements actuels. Comprendre le colobome, c’est déjà se donner les moyens d’agir au mieux pour préserver la vision et la qualité de vie des patients concernés.