Le pityriasis rubra pilaire représente l’une des dermatoses les plus énigmatiques de la pratique dermatologique moderne. Cette affection chronique, caractérisée par une triade sémiologique distinctive, affecte moins d’une personne sur 50 000, rendant son diagnostic particulièrement challengeant pour les praticiens. L’évolution imprévisible de cette pathologie et sa résistance fréquente aux thérapeutiques conventionnelles en font un véritable défi diagnostique et thérapeutique. La compréhension récente des mécanismes physiopathologiques sous-jacents ouvre néanmoins de nouvelles perspectives de prise en charge, notamment avec l’avènement des biothérapies ciblées.
Physiopathologie du pityriasis rubra pilaire : mécanismes kératinocytaires et dysrégulation folliculaire
La compréhension des mécanismes physiopathologiques du pityriasis rubra pilaire a considérablement évolué ces dernières années. Cette dermatose complexe résulte d’une dysrégulation multifactorielle impliquant plusieurs systèmes cellulaires et moléculaires. L’interaction entre facteurs génétiques prédisposants et éléments déclenchants environnementaux semble orchestrer le développement de cette affection chronique.
Anomalies de la différenciation épidermique et hyperprolifération kératinocytaire
L’épiderme des patients atteints présente des anomalies caractéristiques de la différenciation kératinocytaire. Les kératinocytes montrent une hyperprolifération marquée avec un raccourcissement significatif du temps de transit épidermique, passant de 28 jours physiologiques à environ 8-10 jours. Cette accélération du renouvellement cellulaire s’accompagne d’une perturbation de l’expression des protéines de différenciation terminale, notamment la filaggrine et les kératines suprabasales. L’hyperkératose caractéristique résulte directement de ces anomalies, créant l’aspect clinique pathognomonique de la maladie.
Dysfonctionnement de l’unité pilo-sébacée et hyperkératose folliculaire
L’atteinte folliculaire constitue un élément central de la physiopathologie. Les follicules pileux présentent une hyperkératose périfolliculaire massive, créant les papules cornées caractéristiques. Cette hyperkératose résulte d’une dysrégulation de l’homéostasie folliculaire, avec une altération de la desquamation physiologique de l’infundibulum pilaire. Les glandes sébacées associées montrent également des anomalies structurelles et fonctionnelles, contribuant à la xérose cutanée généralisée observée chez ces patients.
Rôle des cytokines pro-inflammatoires IL-1β et TNF-α dans la pathogenèse
L’environnement cytokinique local joue un rôle déterminant dans l’entretien du processus pathologique. L’interleukine-1β et le facteur de nécrose tumorale alpha présentent des taux tissulaires significativement élevés dans les lésions actives. Ces médiateurs pro-inflammatoires stimulent la prolifération kératinocytaire et perturbent les mécanismes de différenciation épidermique normale. La cascade inflammatoire ainsi initiée auto-entretient le processus pathologique, expliquant la chronicité de l’affection et sa tendance aux récidives.
Déficit en vitamine A et perturbation du métabolisme rétinoïde
Bien que controversée
bien documentée, l’hypothèse d’un déficit en vitamine A repose sur plusieurs arguments cliniques et histologiques. On observe en effet des similitudes frappantes entre le pityriasis rubra pilaire et les dermatoses carentielles en vitamine A (phrynodermie), avec des papules folliculaires cornées et une sécheresse cutanée diffuse. Sur le plan biochimique, certains auteurs ont mis en évidence des anomalies d’expression des récepteurs nucléaires aux rétinoïdes (RAR et RXR), suggérant une résistance fonctionnelle plutôt qu’une simple carence absolue. Cette altération du métabolisme rétinoïde expliquerait en partie la bonne réponse de nombreux patients aux rétinoïdes systémiques, malgré des taux sériques de vitamine A souvent normaux.
Classification nosologique de griffiths : typologie clinique et formes héréditaires
Pour mieux appréhender la grande hétérogénéité clinique du pityriasis rubra pilaire, la classification de Griffiths reste aujourd’hui la plus utilisée. Elle distingue six types principaux, fondés sur l’âge de début, l’aspect des lésions, leur extension et l’évolution dans le temps. Cette typologie permet non seulement de décrire plus précisément le tableau clinique, mais aussi d’orienter le pronostic et la stratégie thérapeutique. En pratique, vous entendrez surtout parler des formes de type I à V, les formes associées au VIH constituant un groupe spécifique dans certaines classifications récentes.
Pityriasis rubra pilaire de type I classique de l’adulte
Le type I correspond à la forme classique de l’adulte et représente environ 50 à 60 % des cas de pityriasis rubra pilaire. Il débute le plus souvent entre 40 et 60 ans, chez un sujet auparavant indemne, parfois à la suite d’un épisode infectieux ou d’un stress majeur. L’éruption commence fréquemment au niveau du cuir chevelu ou du tronc, puis s’étend de façon céphalo-caudale pour aboutir à une érythrodermie quasi-généralisée, entrecoupée d’îlots de peau saine. L’évolution se fait habituellement vers une rémission progressive en 2 à 5 ans, ce qui en fait la forme à meilleur pronostic.
Sur le plan thérapeutique, le pityriasis rubra pilaire de type I répond dans la majorité des cas aux rétinoïdes systémiques (acitrétine, isotrénoïne), parfois associés à une photothérapie ou à des dermocorticoïdes puissants. La prise en charge doit toutefois rester individualisée, en tenant compte des comorbidités, de l’âge et du projet de grossesse chez les patientes. Dans cette forme, l’objectif est d’obtenir un blanchiment cutané progressif tout en limitant les effets indésirables, grâce à une surveillance clinique et biologique rapprochée.
Forme atypique de type II et résistance thérapeutique
Le type II, ou forme atypique de l’adulte, se caractérise par une évolution beaucoup plus prolongée, parfois sur plusieurs décennies. Cliniquement, les lésions sont souvent plus épaisses, avec une hyperkératose palmo-plantaire très marquée et douloureuse, pouvant entraîner des fissures profondes et un retentissement fonctionnel important. L’extension est moins volontiers érythrodermique diffuse que dans le type I, mais les plaques érythémato-squameuses sont plus tenaces et moins réversibles. Cette forme touche une proportion plus faible de patients, estimée entre 5 et 10 % des cas.
Le défi majeur du pityriasis rubra pilaire de type II réside dans sa résistance relative aux traitements conventionnels. Les rétinoïdes systémiques, pourtant considérés comme la pierre angulaire de la prise en charge, sont souvent insuffisants ou mal tolérés à long terme. On a ainsi recours plus fréquemment au méthotrexate, à la ciclosporine ou, ces dernières années, aux biothérapies ciblant le TNF-α, l’IL-12/23 ou l’IL-17. Faut-il pour autant parler de “forme réfractaire” d’emblée ? Pas nécessairement, mais il est essentiel d’anticiper une prise en charge prolongée, avec une stratégie thérapeutique séquentielle ou combinée.
Variants juvéniles de types III et IV : formes circonscrites et généralisées
Les types III et IV correspondent aux formes juvéniles du pityriasis rubra pilaire, avec un début avant l’adolescence. Le type III, juvénile classique, se manifeste par une éruption généralisée, souvent rapidement progressive, mimant parfois un psoriasis érythrodermique. L’enfant présente des plaques érythémato-squameuses diffuses, une kératodermie palmo-plantaire et des papules folliculaires typiques. Cette forme, bien que spectaculaire, évolue fréquemment vers une rémission complète en quelques années, surtout lorsqu’un traitement par rétinoïdes oraux est instauré précocement.
À l’inverse, le type IV, ou forme juvénile circonscrite, reste limité à certaines régions, en particulier les genoux, les coudes et parfois le cuir chevelu. Les lésions prennent l’aspect de plaques bien délimitées, orangées, épaisses, pouvant être confondues avec un psoriasis en plaques. La kératodermie palmo-plantaire y est moins constante et la qualité de vie des enfants est souvent moins altérée que dans le type III. Cependant, le risque de persistance à l’âge adulte n’est pas négligeable, ce qui justifie un suivi dermatologique prolongé et régulier.
Type V atypique juvénile et mutations CARD14
Le type V regroupe les formes juvéniles atypiques, souvent familiales, dont l’évolution est chronique et fluctuante. Dans ces formes, les poussées peuvent débuter très tôt dans l’enfance et se prolonger à l’âge adulte, avec des périodes de rémission partielle. Sur le plan clinique, on observe des plaques érythémato-squameuses plus stables, parfois à prédominance acrale, et une kératodermie palmo-plantaire très variable. L’histoire familiale de dermatoses proches (psoriasis, kératodermies héréditaires) doit alerter et conduire à une anamnèse détaillée.
Les progrès de la génétique ont montré que certaines de ces formes juvéniles atypiques sont liées à des mutations du gène CARD14, impliqué également dans certaines formes familiales de psoriasis. Cette découverte illustre la proximité physiopathologique entre ces deux dermatoses et justifie le recours à des biothérapies ciblant les voies IL-23/IL-17 dans les cas sévères. Pour les familles concernées, un conseil génétique peut être proposé, même si le risque de transmission reste globalement faible à l’échelle de la population générale. Là encore, la prise en charge doit être multidisciplinaire, intégrant pédiatres, dermatologues et parfois généticiens.
Sémiologie dermatologique : signes pathognomoniques et diagnostic différentiel
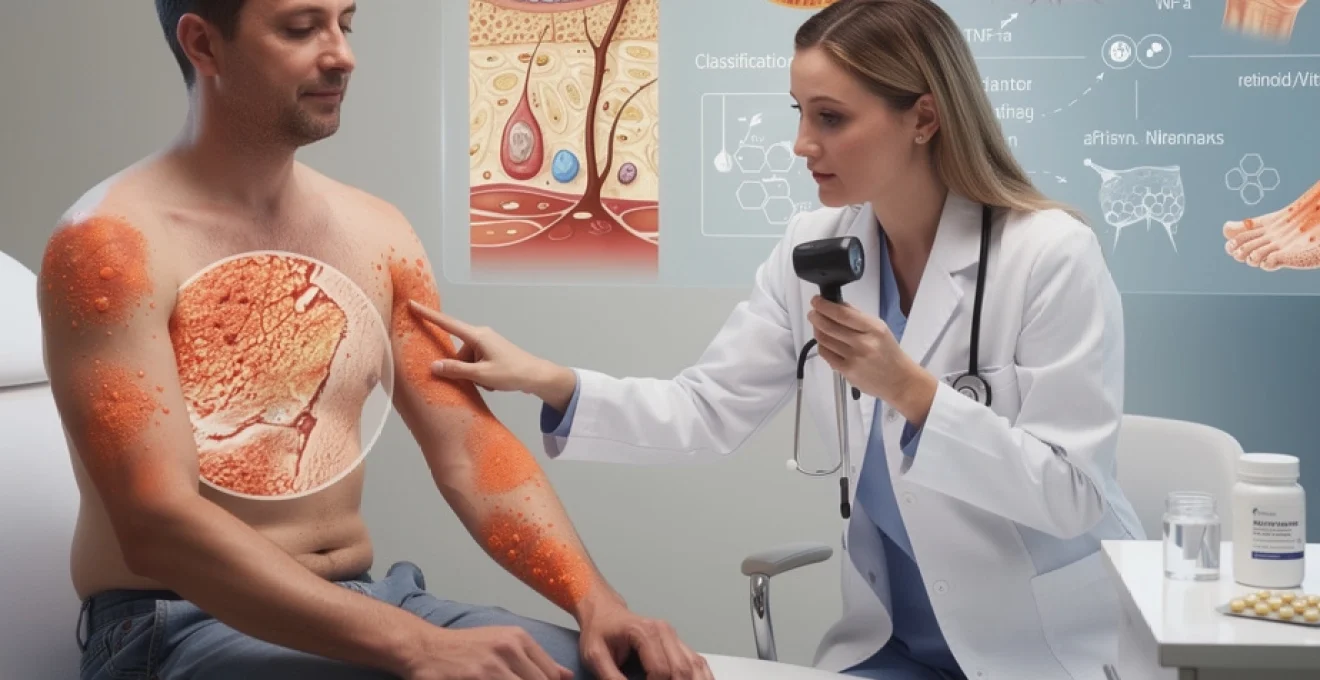
Sur le plan clinique, le pityriasis rubra pilaire présente une sémiologie suffisamment typique pour permettre au clinicien averti de l’identifier, à condition d’y penser. L’association de plaques érythémato-squameuses, de papules folliculaires cornées et d’une kératodermie palmo-plantaire orangée constitue une véritable “signature clinique”. Pourtant, dans les premiers temps, l’affection peut se présenter de façon trompeuse, évoquant un psoriasis ou une dermatite atopique. Comment, dans ce contexte, reconnaître les signes réellement pathognomoniques ?
Érythrodermie avec îlots de peau saine caractéristiques
L’un des éléments les plus évocateurs du pityriasis rubra pilaire est la présence d’une érythrodermie plus ou moins diffuse, parsemée d’îlots de peau saine. Cet aspect en “carte de géographie” crée un contraste net entre les zones enflammées et les plages cutanées indemnes, rarement observé dans d’autres dermatoses inflammatoires. L’extension est souvent céphalique au départ, avec atteinte du cuir chevelu, du visage et du haut du tronc, avant de s’étendre aux membres. La desquamation est fine à modérée, moins farineuse que dans le psoriasis érythrodermique.
Sur le plan symptomatique, le prurit est variable, parfois étonnamment modéré au regard de l’intensité des lésions. Certains patients décrivent plutôt une sensation de brûlure ou de chaleur cutanée, qui peut être majorée en ambiance chaude ou humide. Dans les formes extensives, l’érythrodermie peut s’accompagner de frissons, d’un inconfort thermique important et d’un risque de déshydratation cutanée, justifiant une évaluation clinique attentive. C’est souvent à ce stade que le recours à une hospitalisation devient nécessaire, ne serait-ce que pour assurer une prise en charge globale et sécurisée.
Hyperkératose palmo-plantaire orange et kératodermie punctuée
La kératodermie palmo-plantaire constitue un autre signe majeur, particulièrement utile pour le diagnostic clinique. Les paumes et les plantes présentent un épaississement corné marqué, de coloration jaune-orangé, parfois strié de fissures douloureuses. Cette teinte orangée, proche de celle d’une carotte épluchée, est souvent décrite comme très caractéristique par les dermatologues expérimentés. Elle contraste avec l’aspect plus blanchâtre et nacré des hyperkératoses psoriasiques.
Outre cette hyperkératose diffuse, on peut observer une kératodermie punctuée, faite de multiples petites papules cornées disséminées sur les zones de pression. Ces lésions, en relief, peuvent gêner la marche ou la préhension et impactent fortement la qualité de vie des patients. Une hydratation intensive associée à des kératolytiques (acide salicylique, urée forte concentration) est souvent nécessaire pour assouplir ces zones et limiter les fissures. Vous voyez à quel point une “simple” atteinte palmo-plantaire peut devenir source de handicap au quotidien.
Follicules pilaires kératosiques et papules périfolliculaires
Les papules folliculaires cornées sont l’un des éléments les plus spécifiques du pityriasis rubra pilaire. Il s’agit de petites surélévations rouge-orangé, centrées par un orifice folliculaire obstrué d’un bouchon kératosique. Au toucher, la peau prend un aspect rugueux, comparable au papier de verre fin ou à une chair de poule persistante. Ces papules siègent volontiers sur les faces d’extension des membres, le tronc supérieur et parfois les fesses.
À mesure que la maladie progresse, ces papules peuvent confluer pour former des plaques plus larges, où la composante folliculaire reste néanmoins perceptible à la palpation. Dans certains cas, l’atteinte folliculaire est si marquée qu’elle évoque des kératoses pilaire banales, mais l’association à l’érythrodermie et à la kératodermie palmo-plantaire doit alerter. C’est un peu comme un puzzle : chaque élément pris isolément peut sembler banal, mais leur combinaison dessine un tableau très évocateur lorsqu’on sait le reconnaître.
Diagnostic différentiel avec psoriasis érythrodermique et dermatite atopique
Le diagnostic différentiel du pityriasis rubra pilaire est dominé par le psoriasis, en particulier dans ses formes érythrodermiques, et par certaines dermatites eczématiformes. Le psoriasis érythrodermique présente une rougeur diffuse, souvent plus homogène, sans les îlots de peau saine typiques du PRP. Les squames y sont plus épaisses, blanchâtres, et l’atteinte des ongles (pitting, onycholyse) est plus fréquente. De plus, l’histoire personnelle ou familiale de psoriasis est souvent retrouvée, ce qui oriente le clinicien.
La dermatite atopique sévère peut également prêter à confusion, surtout chez l’enfant, avec un prurit intense et des lésions eczématiformes diffuses. Toutefois, l’absence de kératodermie palmo-plantaire orangée et de papules folliculaires cornées bien individualisées doit faire douter. Les dermatoses kératodermiques héréditaires, certaines ichtyoses ou encore les toxidermies érythrodermiques entrent aussi dans la discussion diagnostique. Dans les situations douteuses, une biopsie cutanée s’impose pour affiner le diagnostic et écarter un psoriasis ou une autre kératinopathie inflammatoire.
Examens paracliniques : histopathologie et biomarqueurs diagnostiques
Si le diagnostic de pityriasis rubra pilaire est avant tout clinique, les examens paracliniques jouent un rôle important pour confirmer l’hypothèse et exclure les diagnostics alternatifs. La biopsie cutanée reste l’examen de référence, particulièrement en cas d’érythrodermie ou de formes atypiques. Elle permet de mettre en évidence un profil histologique assez caractéristique, bien que non totalement spécifique, combinant des anomalies de la kératinisation et un infiltrat inflammatoire dermique discret.
Sur le plan histologique, on retrouve classiquement une hyperkératose orthokératosique avec parakératose focale, une alternance de zones d’hyper et d’hypogranulose, ainsi qu’une acanthose irrégulière. Les orifices folliculaires sont obstrués par des bouchons cornés, corrélats des papules cliniquement observées. Le derme papillaire montre un infiltrat inflammatoire lymphocytaire modéré, associé à une vasodilatation capillaire. Ce “profil mixte” aide à distinguer le PRP d’un psoriasis typique, où la parakératose est plus diffuse et les micro-abcès de Munro plus fréquents.
Au-delà de l’histologie classique, l’ère de la dermatologie de précision voit émerger de nouveaux biomarqueurs potentiels. Des études récentes ont mis en évidence des signatures transcriptomiques spécifiques du pityriasis rubra pilaire, distinctes de celles du psoriasis, notamment concernant l’expression de gènes impliqués dans la kératinisation et la réponse immunitaire innée. À l’avenir, ces outils pourraient permettre de réduire le délai diagnostique, qui dépasse encore 6 à 12 mois dans de nombreux cas. En pratique clinique courante, un bilan biologique standard (NFS, bilan hépatique et rénal, bilan lipidique) est surtout utile pour préparer la mise en route d’un traitement systémique.
Stratégies thérapeutiques : rétinoïdes systémiques et traitements immunosuppresseurs
La prise en charge thérapeutique du pityriasis rubra pilaire reste un véritable défi, en l’absence de recommandations fondées sur de grands essais randomisés. La rareté de la maladie impose de s’appuyer sur des séries de cas, des revues systématiques et l’expérience des centres de référence. Néanmoins, un consensus se dégage autour de plusieurs axes majeurs : soins topiques intensifs, rétinoïdes systémiques en première ligne, puis recours aux immunosuppresseurs classiques ou aux biothérapies en cas de forme sévère ou résistante.
Les rétinoïdes oraux, principalement l’acitrétine chez l’adulte et l’isotrétinoïne chez l’enfant, constituent le traitement de référence des formes étendues. Ils agissent en normalisant la différenciation kératinocytaire et en réduisant l’hyperkératose folliculaire. Les taux de réponse partielle ou complète rapportés avoisinent 60 à 70 %, avec une amélioration visible en quelques semaines à quelques mois. Cependant, ces molécules nécessitent une surveillance rigoureuse : bilan hépatique, profil lipidique, prévention de la tératogénicité chez les femmes en âge de procréer, et prise en charge de la sécheresse cutanéo-muqueuse iatrogène.
En cas d’échec, d’intolérance ou de contre-indication aux rétinoïdes, les traitements immunosuppresseurs prennent le relais. Le méthotrexate, utilisé à faibles doses hebdomadaires, a montré une efficacité intéressante, notamment dans les formes de type II ou associées à des maladies auto-immunes. La ciclosporine peut être proposée en traitement de “sauvetage” pour contrôler rapidement une érythrodermie sévère, avant de relayer vers un traitement de fond mieux toléré. D’autres immunosuppresseurs comme l’azathioprine ou le mycophénolate mofétil sont parfois utilisés en seconde ou troisième ligne, selon le profil du patient.
Les biothérapies ciblant le TNF-α (étanercept, infliximab), l’IL-12/23 (ustékinumab) ou l’IL-17 (sécukinumab) ont profondément modifié le paysage thérapeutique ces dernières années. Bien qu’aucune ne dispose à ce jour d’une AMM spécifique pour le pityriasis rubra pilaire, de nombreuses observations et petites séries suggèrent des taux de réponse élevés, en particulier dans les formes réfractaires aux traitements classiques. Faut-il systématiquement y recourir ? La réponse reste prudente : ces traitements doivent être réservés aux cas sévères, après discussion en RCP, en tenant compte du risque infectieux et du coût. En parallèle, les soins locaux (émollients, kératolytiques, dermocorticoïdes) et la photothérapie UVB à spectre étroit gardent toute leur place pour optimiser le confort cutané.
Pronostic évolutif et surveillance dermatologique à long terme
Le pronostic du pityriasis rubra pilaire dépend étroitement du type clinique, de l’âge de début et de la précocité de la prise en charge. Les formes classiques de l’adulte (type I) et certaines formes juvéniles généralisées ont tendance à s’amender progressivement en quelques années, parfois avec une rémission complète et durable. À l’inverse, les formes atypiques de l’adulte (type II) et les formes juvéniles liées à des mutations CARD14 présentent une évolution plus chronique, avec des poussées récurrentes et un risque de persistance à long terme. L’espérance de vie n’est toutefois pas réduite par la maladie elle-même, ce qui est un point rassurant à rappeler aux patients.
La surveillance dermatologique à long terme vise plusieurs objectifs : évaluer le contrôle des symptômes, adapter la stratégie thérapeutique, dépister les effets indésirables des traitements systémiques et accompagner le patient dans la gestion de sa qualité de vie. Un suivi tous les 3 à 6 mois est généralement recommandé pour les patients sous rétinoïdes, méthotrexate ou biothérapies, avec un bilan biologique régulier. En dehors des phases actives, une consultation annuelle peut suffire, à condition que le patient soit bien informé des signes d’alerte justifiant une réévaluation rapide (aggravation soudaine, fièvre, signes de surinfection).
Au-delà des aspects strictement médicaux, le pityriasis rubra pilaire peut avoir un impact psychologique important, comparable à celui observé dans le psoriasis sévère. Les lésions visibles, la gêne fonctionnelle et la chronicité de la maladie peuvent altérer l’estime de soi et la vie sociale. C’est pourquoi une approche globale, intégrant éventuellement un soutien psychologique et une orientation vers des associations de patients, est vivement conseillée. En somme, reconnaître et traiter cette dermatose rare, c’est aussi apprendre à accompagner les patients dans la durée, en tenant compte de leurs attentes, de leurs contraintes et de leurs projets de vie.